Née en France avec la loi du 4 janvier 1993, l’hémovigilance est un élément de la sécurité transfusionnelle et a pour objet la surveillance, l'évaluation et la prévention des incidents et des effets indésirables survenant chez les donneurs ou les receveurs de produits sanguins labiles (PSL). Ce système est basé sur une organisation stricte entre les différents acteurs à des niveaux différents avec des tâches spécifiques à réaliser.
Organisation de l'hémovigilance
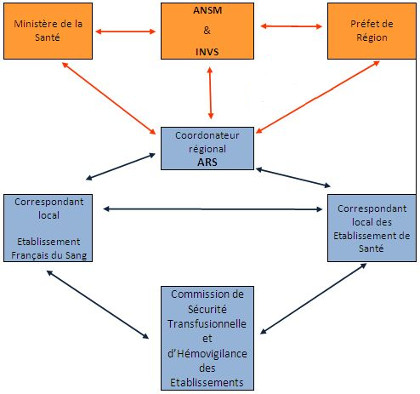
Rôle de l'Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM)
L'ANSM assure la mise en oeuvre de l'hémovigilance. Elle en définit les orientations, anime et coordonne les actions des différents intervenants et veille au respect des procédures de surveillance organisées par la présente section. Elle prend, le cas échéant, les mesures appropriées en vue d'assurer la sécurité transfusionnelle ou saisit les autorités compétentes.
Rôle des Coordonnateurs Régionaux d'Hémovigilance (CRH)
Placé auprès de l'Agences Régionales de Santé (ARS), le CRH est chargé de :
- Suivre la mise en oeuvre des dispositions de l’hémovigilance, des décisions de l'ANSM et des actions entreprises par les Comités de Sécurité Transfusionnelle et d'Hémovigilance des établissements de soins (CSTH)
- Entretenir des relations directes avec chacun des correspondants d'hémovigilance de la région, de veiller avec eux à la qualité et à la fiabilité des informations recueillies et de se tenir informé de toute difficulté que les correspondants rencontreraient dans l'exercice de leur mission
- Informer le préfet de région et l'agence de son activité, notamment par un rapport annuel d'activité
- Proposer, le cas échéant à l'agence, l'adoption de toute mesure susceptible d'améliorer la qualité, la fiabilité et la cohérence du dispositif d'hémovigilance
- Saisir sans délai le préfet de région et l'agence de toute difficulté susceptible de compromettre la sécurité transfusionnelle et d'en informer simultanément l'Etablissement Français du Sang
- Proposer, le cas échéant, au préfet de département les mesures à prendre au vu des fiches de déclarations.
Rôle des correspondants d'hémovigilance des ETS
Le correspondant d'hémovigilance des Etablissements de Transfusions Sanguines (ETS) est chargé d'assurer :
- Le recueil et la conservation des informations, en veillant à leur qualité et à leur fiabilité
- La déclaration de tout effet indésirable grave survenu chez un donneur de sang et de tout effet indésirable survenu chez un receveur de PSL ainsi que de tout incident grave
- La communication des informations à l'ANSM, à l’InVS et au CRH
- L'information des établissements de soins sur l'usage des PSL distribués ou délivrés par son ETS référent et la transmission à ces établissements de ces informations
- Le signalement à l'ANSM et au CRH de toute difficulté susceptible de compromettre la sécurité transfusionnelle
- Les investigations à entreprendre en cas d'urgence suite à des effets indésirables ou des incidents graves. Dans ce cas, il informe sans délai l'ANSM, qui décide de la poursuite ou de l'interruption de ces investigations, ainsi que le CRH.
Rôle des correspondants d'hémovigilance des Etablissements de Santé (ES)
Le correspondant d'hémovigilance ES est chargé d'assurer :
- La déclaration de tout effet indésirable survenu chez un receveur de produits sanguins labiles ainsi que de tout incident grave
- Le recueil et la conservation des informations, en veillant à la qualité et à la fiabilité de ces informations
- La communication à l'ANSM et au CRH des informations qu'ils sollicitent
- La transmission à l'ETS référent des informations
- Le signalement à l'ANSM et au CRH de toute difficulté susceptible de compromettre la sécurité transfusionnelle
- Les investigations à entreprendre en cas d'urgence à la suite des effets indésirables survenus chez les receveurs de produits sanguins labiles ou des incidents graves. Dans ce cas, il informe sans délai le CRH, qui décide de la poursuite ou de l'interruption de ces investigations, et l'ANSM.
Signalement d'un effet indésirable receveur ou donneur
Le signalement et la déclaration des incidents graves, des effets indésirables graves survenus chez un donneur de sang (EID) et des effets indésirables survenus chez un receveur de PSL (EIR) sont obligatoires. Les fiches de déclaration doivent être adressées simultanément à l’ANSM et au CRH (coordonnateur régional d'hémovigilance). L'EFS et le CTSA sont également destinataires des fiches de déclaration les concernant.
Un incident grave est un accident ou une erreur lié au prélèvement de sang, à la qualification biologique du don, à la préparation, à la conservation, à la distribution, à la délivrance ou à l’utilisation de produits sanguins labiles, susceptible d’affecter la sécurité ou la qualité de ce produit et d’entraîner des effets indésirables graves. C’est-à-dire des effets indésirables entraînant la mort ou mettant la vie en danger, entraînant une invalidité ou une incapacité, ou provoquant ou prolongeant une hospitalisation ou tout autre état morbide. Les incidents graves doivent être déclarés sur une Fiche d'Incident Grave (FIG).
Effet indésirable
Les EI sont classés selon les critères de gravité suivants :
Grade 1 : absence de menace vitale immédiate ou à long terme
Grade 2 : morbidité à long terme
Grade 3 : menace vitale immédiate
Grade 4 : décès du receveur.
Et selon les critères d’imputabilité suivants :
Imputabilité 4 : certaine (les bilans prouvent l’origine transfusionnelle de l’effet indésirable)
Imputabilité 3 : vraisemblable (l’effet indésirable ne semble pas pouvoir être expliqué par une cause intercurrente, et il est retenu des éléments d’orientation en faveur de l’origine transfusionnelle de l’incident)
Imputabilité 2 : possible (l’effet indésirable pourrait être expliqué soit par une origine transfusionnelle soit par une cause intercurrente sans qu’il soit possible de trancher en l’état de l’enquête)
Imputabilité 1 : douteuse (l’effet indésirable ne semble pas pouvoir être complètement expliqué par l’administration du produit sanguin labile, sans qu’on puisse totalement l’exclure)
Imputabilité 0 : exclue (la preuve a été faite que le produit sanguin labile n’est pas en cause dans la survenue de l’effet indésirable)
Les effets indésirables receveurs doivent être déclarés sur une Fiche EIR ou EID.